Note
Go to the end to download the full example code
SARI - By specimen w/ sns.heatmap
Todo
Explain…
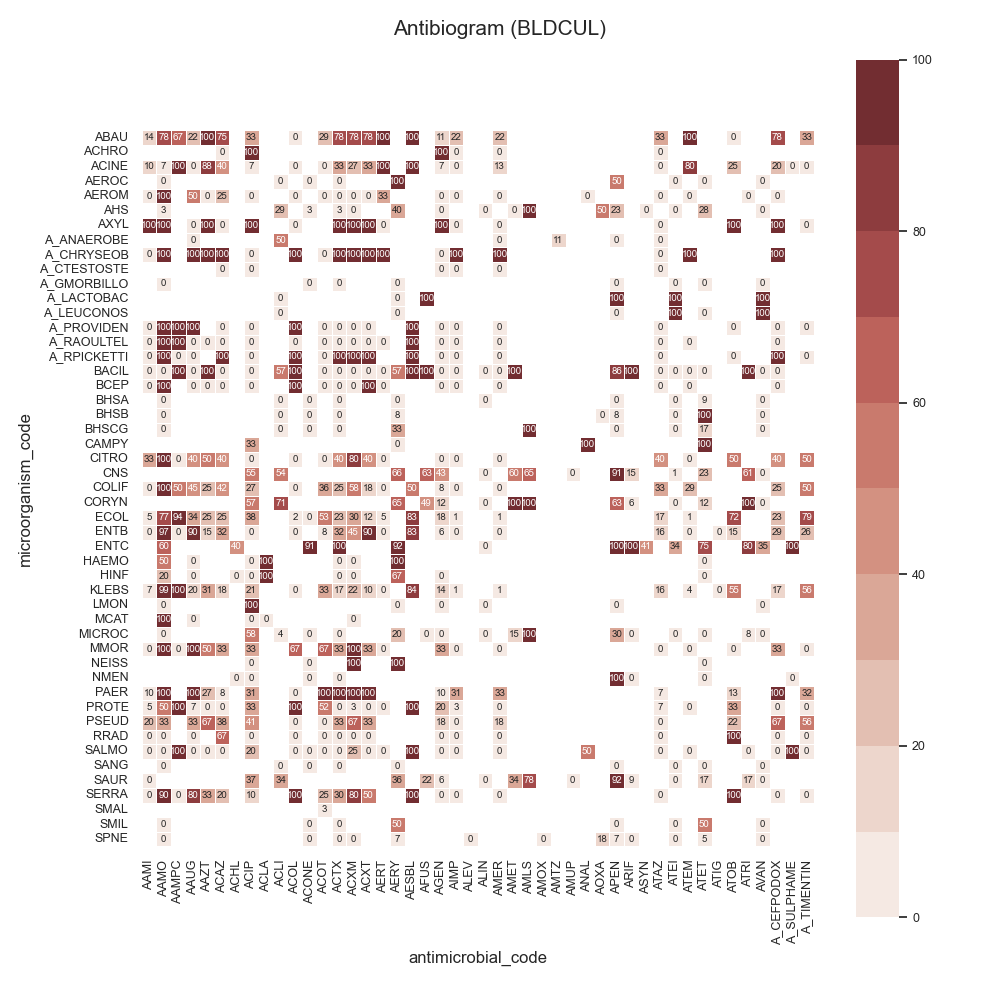
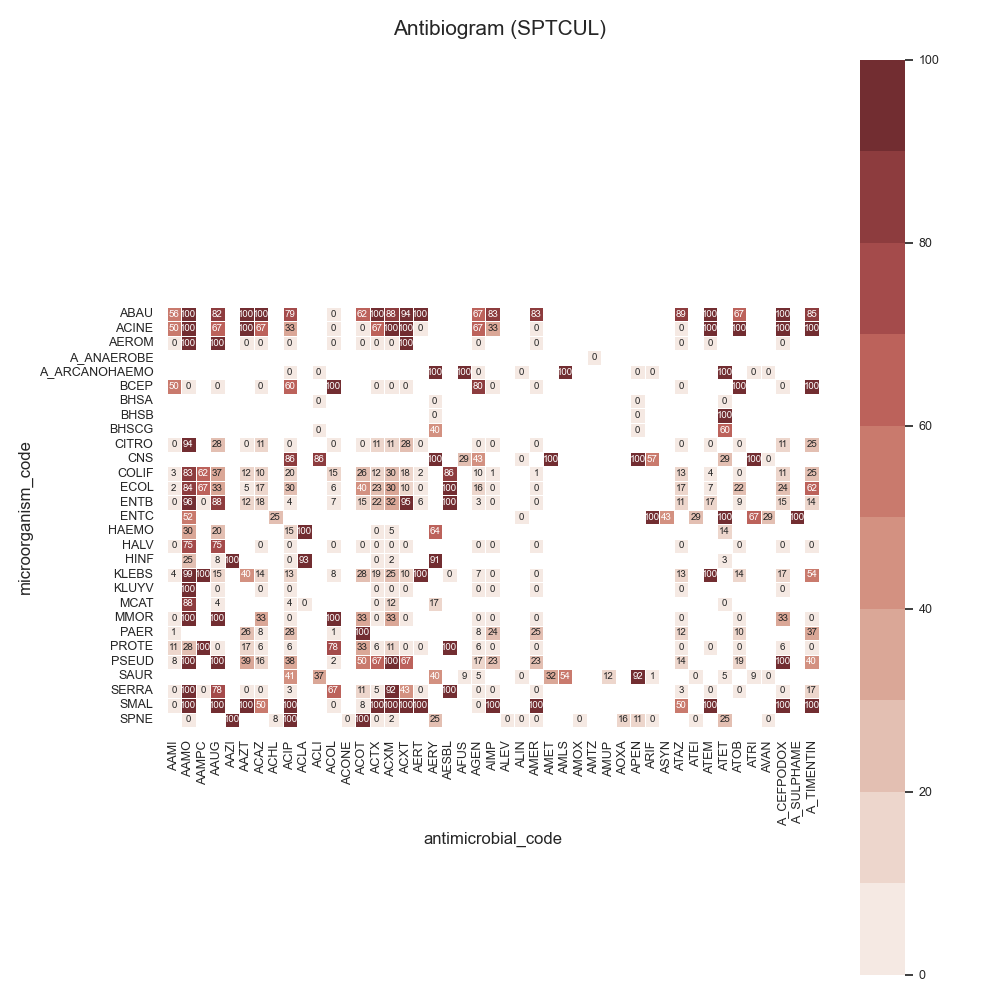
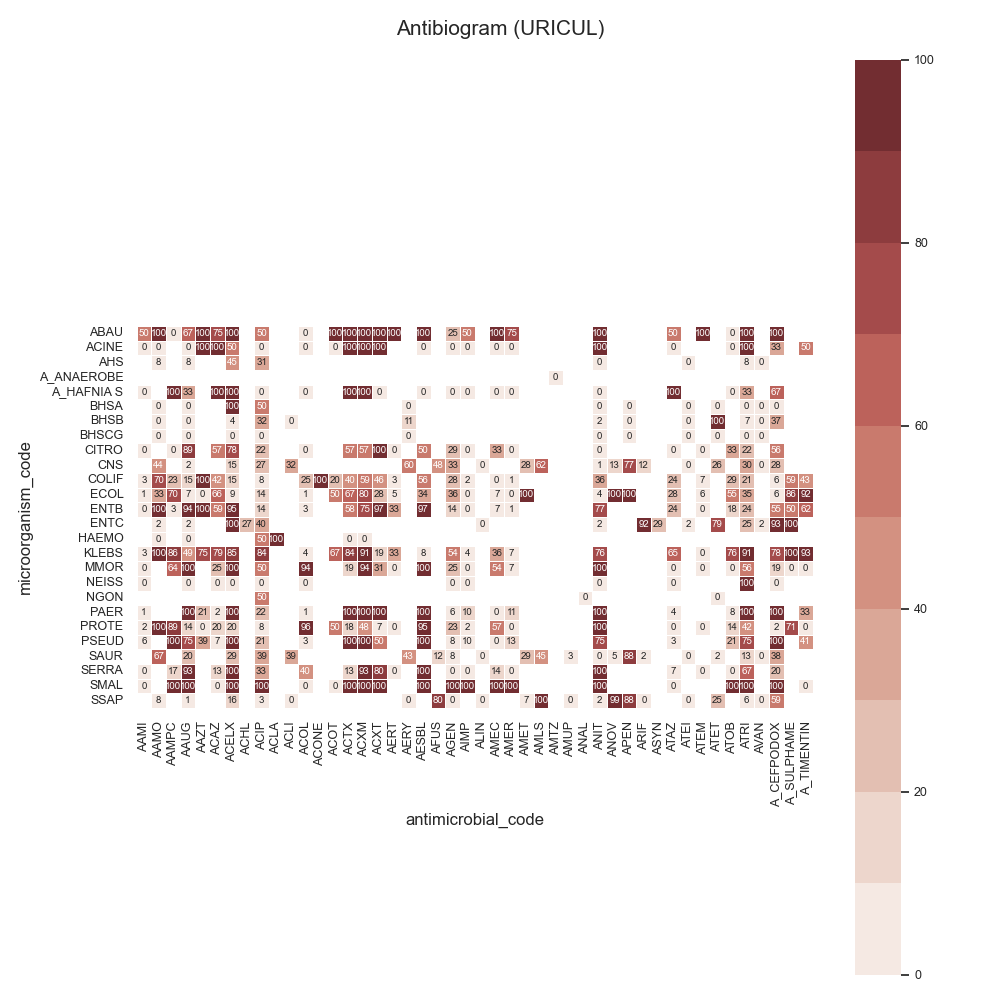
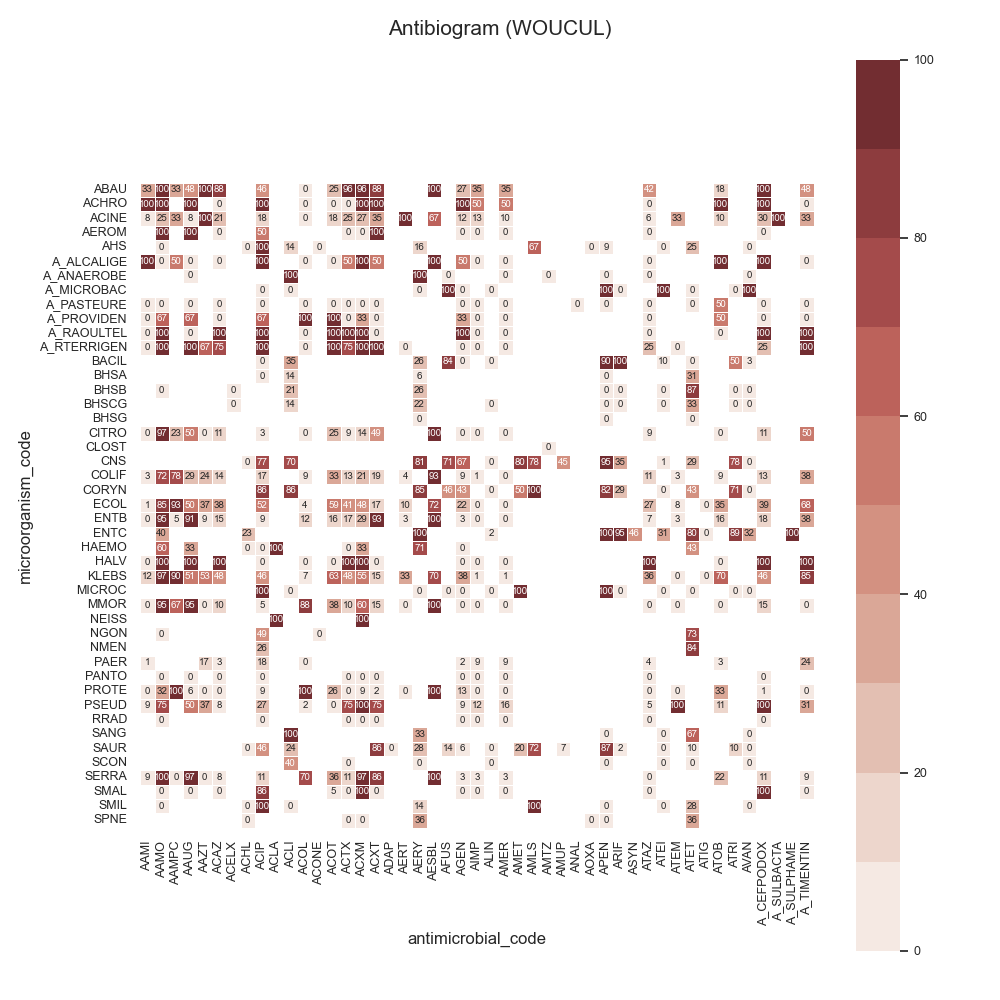
Data:
date_received date_outcome patient_id laboratory_number specimen_code specimen_name specimen_description ... microorganism_name antimicrobial_code antimicrobial_name sensitivity_method sensitivity mic reported
0 2009-01-03 NaN 20091 X428501 BLDCUL NaN blood ... klebsiella AAMI amikacin NaN sensitive NaN NaN
1 2009-01-03 NaN 20091 X428501 BLDCUL NaN blood ... klebsiella AAMO amoxycillin NaN resistant NaN NaN
2 2009-01-03 NaN 20091 X428501 BLDCUL NaN blood ... klebsiella AAUG augmentin NaN sensitive NaN NaN
3 2009-01-03 NaN 20091 X428501 BLDCUL NaN blood ... klebsiella AAZT aztreonam NaN sensitive NaN NaN
4 2009-01-03 NaN 20091 X428501 BLDCUL NaN blood ... klebsiella ACAZ ceftazidime NaN sensitive NaN NaN
... ... ... ... ... ... ... ... ... ... ... ... ... ... .. ...
319117 2009-12-31 NaN 24645 H2012337 BLDCUL NaN blood ... enterococcus AAMO amoxycillin NaN sensitive NaN NaN
319118 2009-12-31 NaN 24645 H2012337 BLDCUL NaN blood ... enterococcus ALIN linezolid NaN sensitive NaN NaN
319119 2009-12-31 NaN 24645 H2012337 BLDCUL NaN blood ... enterococcus ASYN synercid NaN resistant NaN NaN
319120 2009-12-31 NaN 24645 H2012337 BLDCUL NaN blood ... enterococcus ATEI teicoplanin NaN sensitive NaN NaN
319121 2009-12-31 NaN 24645 H2012337 BLDCUL NaN blood ... enterococcus AVAN vancomycin NaN sensitive NaN NaN
[319122 rows x 15 columns]
Columns:
Index(['date_received', 'date_outcome', 'patient_id', 'laboratory_number', 'specimen_code', 'specimen_name', 'specimen_description', 'microorganism_code', 'microorganism_name', 'antimicrobial_code', 'antimicrobial_name', 'sensitivity_method', 'sensitivity', 'mic', 'reported'], dtype='object')
SARI (overall):
intermediate resistant sensitive freq sari
specimen_code microorganism_code antimicrobial_code
BFLCUL AHS ACHL 0.0 0.0 1.0 1.0 0.0000
ACLI 0.0 0.0 2.0 2.0 0.0000
ACTX 0.0 0.0 1.0 1.0 0.0000
AERY 0.0 1.0 1.0 2.0 0.5000
APEN 0.0 0.0 2.0 2.0 0.0000
... ... ... ... ... ...
XINCUL SAUR ATEI 0.0 0.0 1365.0 1365.0 0.0000
ATET 0.0 67.0 1288.0 1355.0 0.0494
ATRI 0.0 145.0 1213.0 1358.0 0.1068
AVAN 0.0 0.0 1364.0 1364.0 0.0000
SMAL ACOT 0.0 0.0 8.0 8.0 0.0000
[4491 rows x 5 columns]
Cultures:
specimen_code
URICUL 116627.0
WOUCUL 94918.0
XINCUL 21427.0
SPTCUL 21113.0
BLDCUL 20333.0
ENTCUL 13110.0
T&FCUL 8150.0
MRSCUL 7865.0
VAGCUL 7425.0
EYECUL 2839.0
GUMCUL 1634.0
FAECUL 1317.0
URECUL 802.0
TISCUL 474.0
BFLCUL 450.0
SEMCUL 290.0
NEOCUL 213.0
PDFCUL 68.0
CSFCUL 32.0
RGNS 20.0
FUNSTC 14.0
TBCUL 1.0
Name: freq, dtype: float64
9 # Libraries
10 import sys
11 import numpy as np
12 import pandas as pd
13 import seaborn as sns
14 import matplotlib as mpl
15 import matplotlib.pyplot as plt
16
17 # Import specific libraries
18 from pyamr.core.sari import SARI
19 from pyamr.core.freq import Frequency
20 from pyamr.datasets.load import make_susceptibility
21
22 # -------------------------
23 # Configuration
24 # -------------------------
25 # Configure seaborn style (context=talk)
26 sns.set(style="white")
27
28 # Set matplotlib
29 mpl.rcParams['xtick.labelsize'] = 9
30 mpl.rcParams['ytick.labelsize'] = 9
31 mpl.rcParams['axes.titlesize'] = 11
32 mpl.rcParams['legend.fontsize'] = 9
33
34 # Pandas configuration
35 pd.set_option('display.max_colwidth', 40)
36 pd.set_option('display.width', 300)
37 pd.set_option('display.precision', 4)
38
39 # Numpy configuration
40 np.set_printoptions(precision=2)
41
42
43 # -------------------------------------------
44 # Load data
45 # -------------------------------------------
46 # Load data
47 data = make_susceptibility()
48
49 # Show
50 print("\nData:")
51 print(data)
52 print("\nColumns:")
53 print(data.columns)
54
55 # -------------------------------------------
56 # Compute SARI
57 # -------------------------------------------
58 # Libraries
59 from pyamr.core.sari import SARI
60
61 # Create sari instance
62 sari = SARI(groupby=['specimen_code',
63 'microorganism_code',
64 'antimicrobial_code',
65 'sensitivity'])
66
67 # Compute SARI overall
68 sari_overall = sari.compute(data,
69 return_frequencies=True)
70
71 # Show
72 print("SARI (overall):")
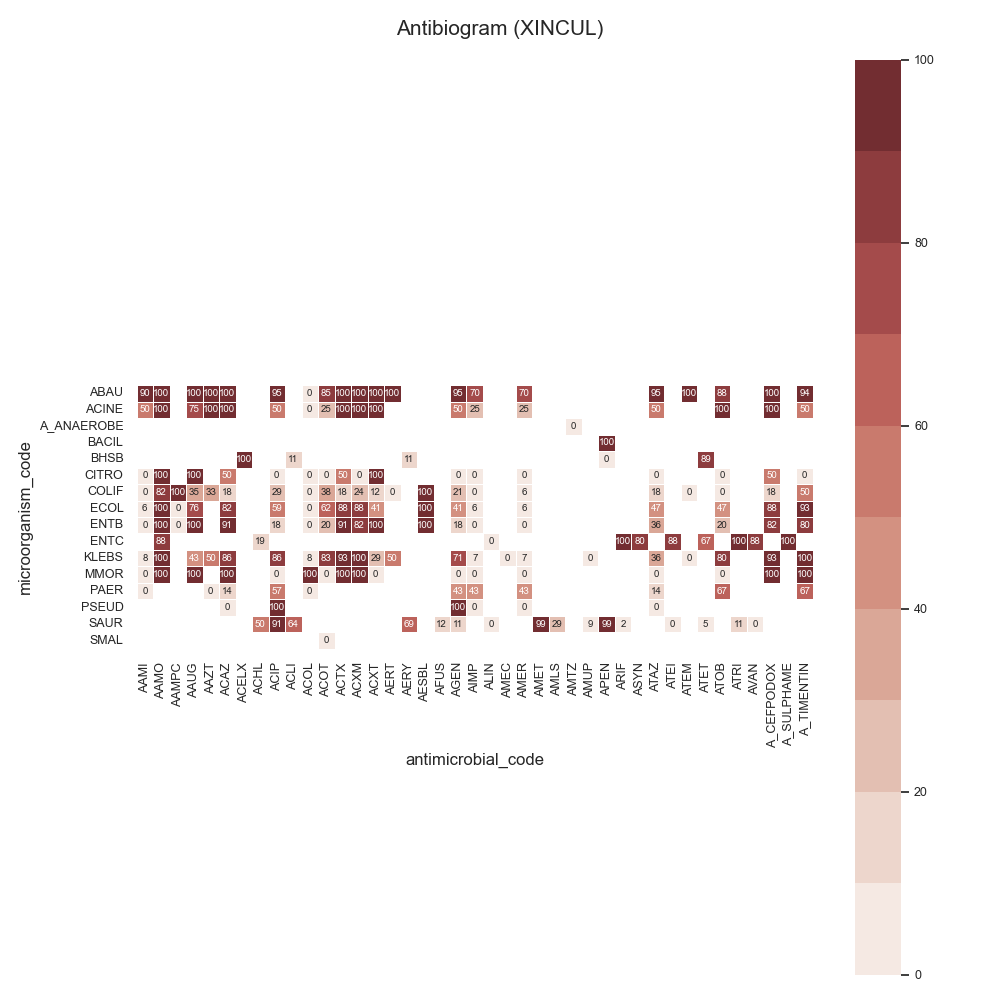
73 print(sari_overall)
74
75 # -------------------------------------------
76 # Plot
77 # -------------------------------------------
78 # Reset
79 sari_overall = sari_overall.reset_index()
80
81 # Count records per specimen
82 specimen_count = sari_overall \
83 .groupby('specimen_code').freq.sum() \
84 .sort_values(ascending=False)
85
86 # Show
87 print("Cultures:")
88 print(specimen_count)
89
90 # Filter
91 sari_overall = sari_overall[sari_overall \
92 .specimen_code.isin( \
93 specimen_count.index.values[:5])]
94
95 # Loop
96 for specimen, df in sari_overall.groupby(by='specimen_code'):
97
98 # -------------
99 # Create matrix
100 # -------------
101 # Filter
102 matrix = df.copy(deep=True)
103 matrix = df.reset_index()
104 #matrix = matrix[matrix.freq > 100]
105
106 # Pivot table
107 matrix = pd.pivot_table(matrix,
108 index='microorganism_code',
109 columns='antimicrobial_code',
110 values='sari')
111
112 # ------------
113 # Plot Heatmap
114 # ------------
115 # Create figure
116 f, ax = plt.subplots(1, 1, figsize=(10, 10))
117
118 # Create colormap
119 cmap = sns.color_palette("Reds", desat=0.5, n_colors=10)
120
121 # Specify cbar axes
122 # cbar_ax = f.add_axes([.925, .3, .05, .3])
123
124 # Plot
125 ax = sns.heatmap(data=matrix*100, annot=True, fmt=".0f",
126 annot_kws={'fontsize': 7}, cmap=cmap,
127 linewidth=0.5, vmin=0, vmax=100, ax=ax,
128 #cbar_ax=cbar_ax,
129 xticklabels=1, yticklabels=1)
130
131 # Configure axes
132 ax.set(aspect="equal")
133
134 # Set rotation
135 plt.yticks(rotation=0)
136
137 # Add title
138 plt.suptitle("Antibiogram (%s)" % specimen, fontsize=15)
139
140 # Tight layout
141 plt.tight_layout()
142
143 # Show
144 plt.show()
Total running time of the script: ( 0 minutes 4.514 seconds)